This is an archival copy of the Visualization Group's web page 1998 to 2017. For current information, please vist our group's new web page.
|
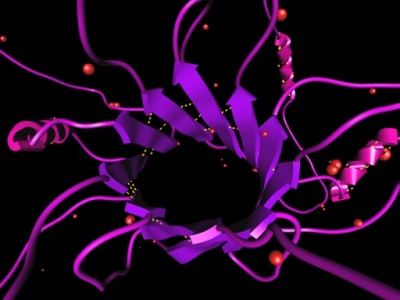
One of the grand challenges is the protein structure prediction problem. The problem consists of determining tertiary structure of a protein given its primary sequence of amino acids. Experimental approaches, like X-ray crystallography and NMR spectroscopy, are very time consuming. Computer simulations that predict the protein fold are a promising alternative. Computational techniques that aim to solve tertiary structure conformation will not only tackle the folding of existing proteins but also the folding of "engineered" proteins, including those designed for drug purposes. Two kinds of methods have been identified to tackle the protein structure prediction problem: knowledge-based methods that rely on the presence of homologous proteins (in sequence or structure) in the databases, and physics-based methods that emphasize more the physical principles. Although the physics-based methods are extremely important to find genuine new folds, they are also extremely expensive. Thus, knowledge-based methods are a valid alternative when there is some homology. However, because in most cases only fragments of the proteins are similar, the results achieved by these methods may be limited. This image shows a protein molecule crafted and visualized with the ProteinShop software, developed at LBNL. More information. |