This is an archival copy of the Visualization Group's web page 1998 to 2017. For current information, please vist our group's new web page.
Meeting Challenges in Molecular Biology through Interactive Visual Data Analysis
|
Introduction
ProteinShop is a software application that facilitates solutions to the protein prediction problem through a combination of interactive features and visualization capabilities.
- It helps researchers automatically generate tertiary structures from data available from prediction servers accessible via the Internet.
- It enables scientists to apply their biochemical knowledge and intuition during interactive manipulation of protein structures.
- It provides visualization of free energy computed during modeling to facilitate comparison and analysis of alternative structures.
- It accelerates the discovery of low-energy configurations by applying local optimizations to user-selected protein structures.
- It allows steering of remotely running global optimization code, decreasing the time required to finish.
History
Members of our research group at Lawrence Berkeley National Laboratories developed a physics-based protein structure prediction method using a sophisticated global optimization approach. The high computational cost of this method motivated the development of ProteinShop to increase the method's efficiency by providing interactive front end systems at different stages of the global optimization process.
ProteinShop was instrumental to our team's participation in the Fifth and Sixth Critical Assessments of Techniques for Protein Structure Prediction (CASP5 in 2002, and CASP6 in 2004). The total time required to produce a set of initial configurations for the global optimization method was reduced from the days it took during CASP4 in 2000 to a matter of hours during CASP5 and CASP6, substantially increasing the number of targets that our team was able to submit. ProteinShop's capabilities also allowed us to attempt targets of far greater size and topological complexity during CASP5 and CASP6 than we could attempt during CASP4.
The following screen shots show initial configurations for the CASP6 targets T227, T230, and T239, respectively.
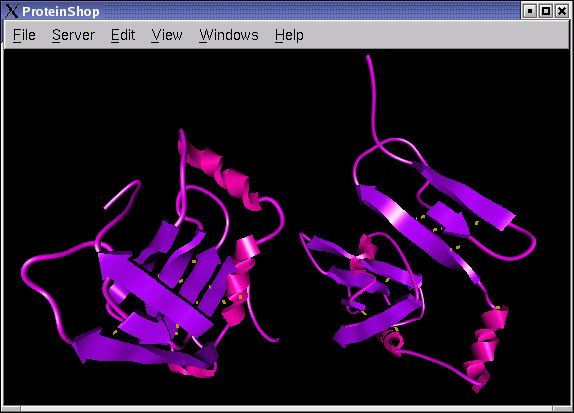
|
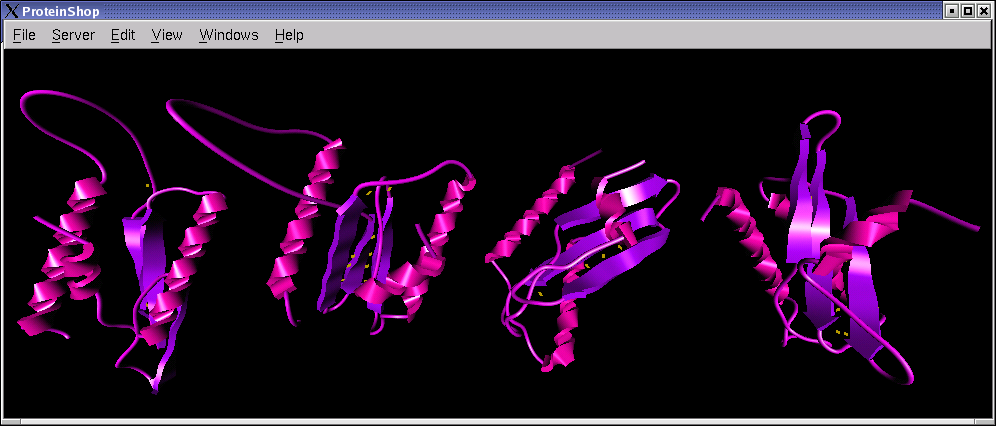
|
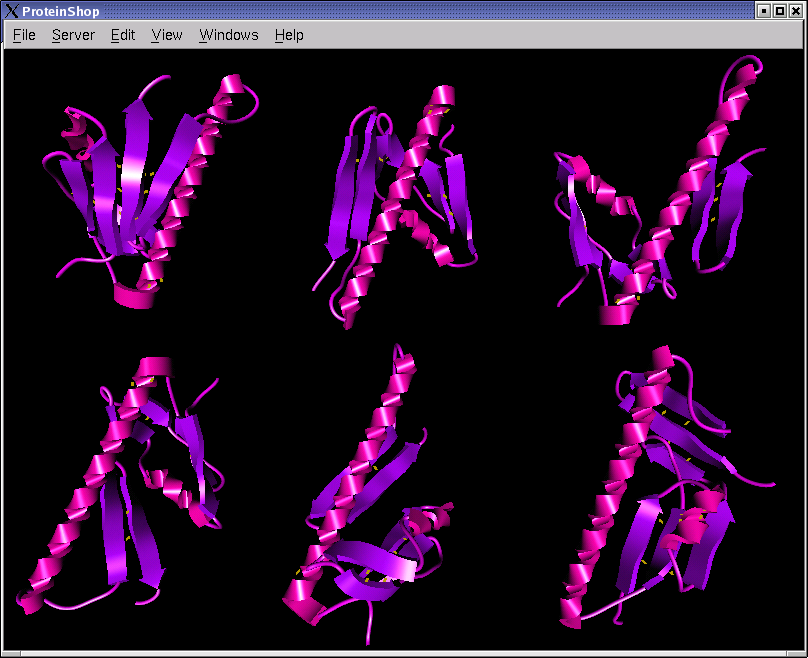
|
Description
From an input sequence of amino acid residues and secondary structure predictions, ProteinShop generates an initial geometric configuration for the molecule. The user interactively reshapes the molecule using a set of geometric tools, trying different arrangements and packings of the secondary structure elements. From these configurations the user selects those with the lowest energy values. ProteinShop's energy visualization lets the user quickly evaluate the stability of each arrangement, rapidly developing a number of candidate configurations for input to the global optimization algorithm.
ProteinShop's interface helps users steer the global optimization process and guide the search through the vast conformation space with human knowledge and intuition. To this end, the user can visualize and manipulate the structures created by the global optimization process as it runs on a remote supercomputer. Once modified by the user, the structures need to be locally optimized before they are placed back in the global optimization process for further reduction. This unique combination of intuitive human knowledge and supercomputing power produces optimized protein structures more quickly than ever before.
Finding an accurate energy function is one of the major concerns in the protein folding community. ProteinShop's energy visualization features help scientists study energy functions by visualizing the effect of the different components in the folding process.
Recent Work
ProteinShop now allows multiple proteins to be manipulated in the same environment. This allows comparative analysis of adjoining tertiary structures during the interactive manipulation process.
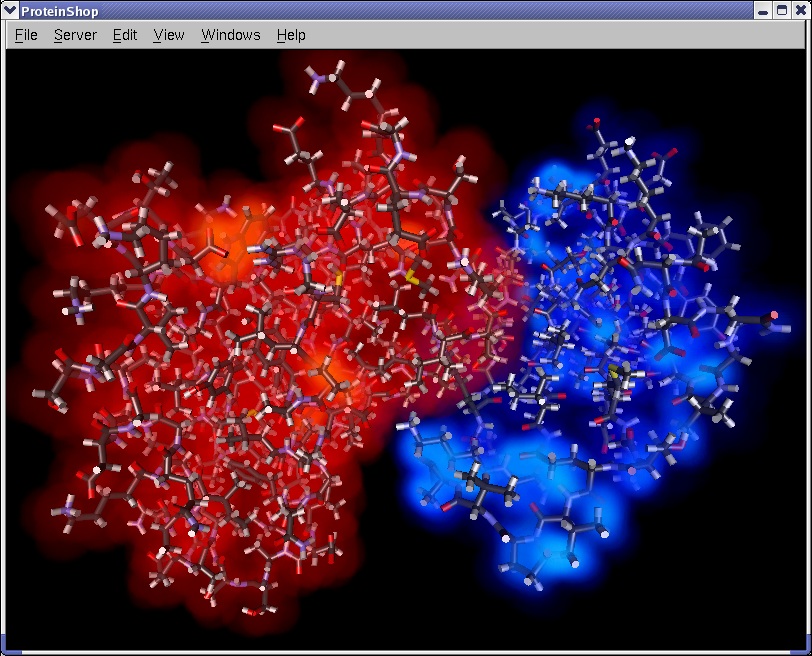
|
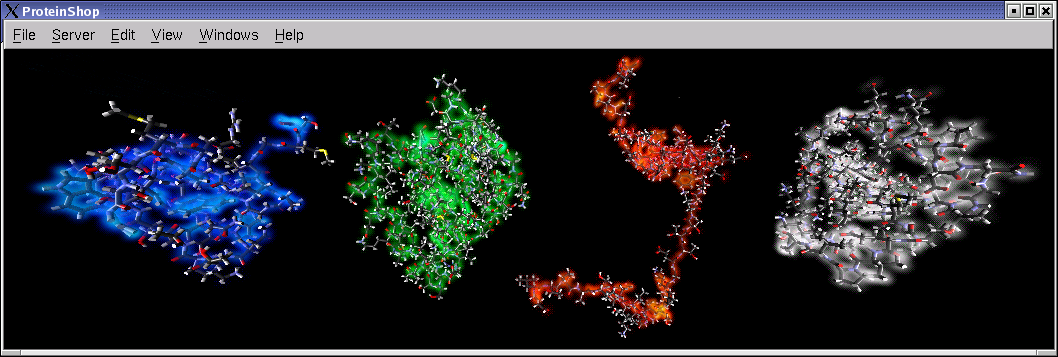
The following screen shot illustrates the energy visualization features in ProteinShop. Glowing clusters represent amino acid residues that contribute most of the total energy value. Red balls represent atoms that are least stable, and green balls represent atoms that are most stable.
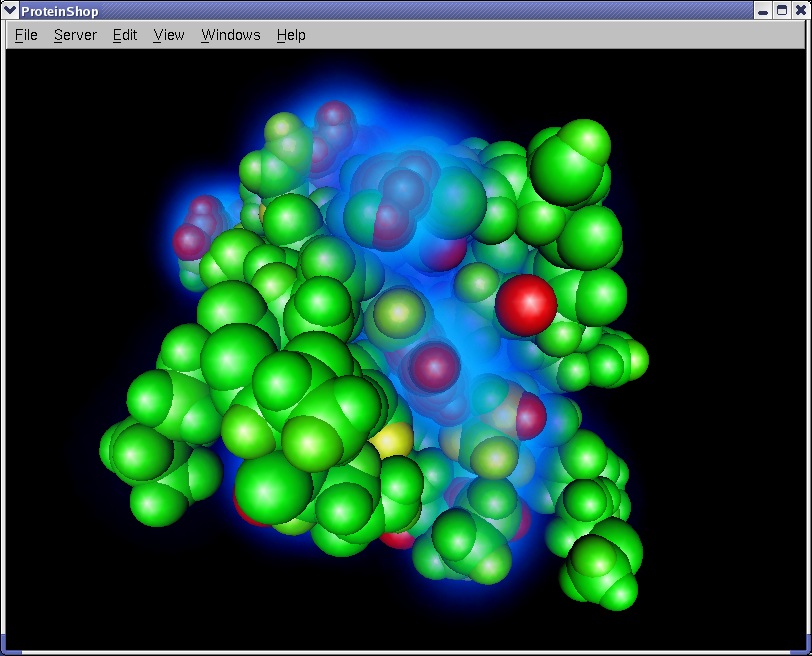
|
The following movie shows the bonds and energies of T209 from CASP6 over the course of the local energy minimization procedure at a rate of one frame for every 20 simulation steps. This movie was generated by ProteinShop using its energy visualization features during the minimization run. Atoms with low energy derivatives are invisible. Atoms and residues that glow brightly have large energy derivatives. The backbone is visible as a continuous chain of dimmer, but visible, atoms.
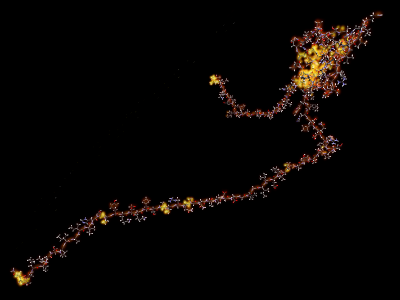
|
Future Work
The energy visualization quality depends on the colors and opacities of the elements, which in turn comes from the transfer functions. As protein energy is minimized, the distribution of energy terms can change so much that the transfer function initially selected no longer offers much information. Adaptive transfer functions will analyze the protein during minimization, automatically adjusting to highlight newly emergent features of the data. Additional classifications will also be implemented; for example, it will be possible to distinguish between hydrophobic and hydrophilic residues in the energy rendering.
References
- S. Crivelli, O. Kreylos, B. Hamann, N. Max, and W. Bethel. "ProteinShop: A tool for interactive protein manipulation and steering." Journal of Computer-Aided Molecular Design. Volume 18 (2004), pp. 271-285.
- O. Kreylos, N. Max, B. Hamann, S. Crivelli, and W. Bethel. "Interactive Protein Manipulation" IEEE Visualization. 2003, pp. 581-588.
- S. Crivelli, E. Eskow, B. Bader, V. Lamberti, R. Byrd, R. Schnabel, and T. Head-Gordon. "A Physical Approach to Protein Structure Prediction". Biophysics Journal. Volume 82 (2002), pp. 36-49.
- C. Crawford, W. Bethel, B. Hamann, O. Kreylos, N. Max, R. Oliva, and S. Crivelli. "Visualization of Energy Optimization" (poster). SIAM Conference on the Life Sciences, 2004.
- Download: ProteinShop download (sourceforge).